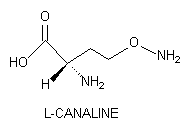
L-Canaline, the L-2-amino-4-(aminooxy)butyric acid structural analog of L-ornithine is a powerful antimetabolite stored in many leguminous plants. This nonprotein amino acid reacts vigorously with the pyridoxal phosphate moiety of vitamin B6-containing enzymes to form a covalently-bound oxime that inactivates, often irreversibly, the enzyme. Canaline is not only capable of inhibiting ornithine-dependent enzymic activity, but it also can function as a Iysine antagonist. Recently, this natural product was found to possess significant antineoplastic in vitro activity against human pancreatic cancer cells.
NATURAL OCCURRENCE
L-Canaline, the L-2-amino-4-(aminooxy)butyric acid structural analog of L-ornithine, is biosynthesized by arginase (EC 3.2.1.5)-mediated hydrolysis of L- Canavanine, an arginine antimetabolite of leguminous plants [Scheme 1] (1,2). Canaline is unique in being the only naturally occurring amino acid possessing an aminooxy moiety.
Because arginase directs the conversion of arginine to ornithine and urea, as part of the KrebsHenseleit ornithine-urea cycle, this enzyme appears to be widely, perhaps universally distributed in higher plants (3). Thus, any canavanine-storing legume can synthesize canaline.
ARTICLE OF HUMAN DIET
At present, only a few canavanine-containing seeds are articles of human diet, but this will undoubtedly change as world demand for protein increases. Jack bean, Canavalia ensiformis and sword bean, Canavalia gladiata are widely eaten, particularly in Asia and parts of the tropics and Africa. In ancient times, seeds of C. ensiformis were an important nitrogen source in the New World (4). These legumes grow productively in marginal habitats, especially in tropical lowlands with depleted soils and sub-optimal climatic conditions. Production yields of 4,600 kg/ha have been obtained with C. ensiformis-about five times the average yield for pulses in the tropics (5).
In North America, the most widely eaten canavanine-containing plant is undoubtedly alfalfa, Medicago saliva. Canavanine is the principal nonprotein amino acid of the seed and it is found in the young sprout as well (6,7). Recent determinations in my laboratory of the natural abundance of canavanine in commercially-available alfalfa sprouts discloses canavanine levels as high as 2.6% of the dry matter.
Studies of canavanine metabolism in the rat reveals that canaline formation from canavanine is the preponderant reaction pathway for canavanine catabolism (8). Thus, it is reasonable to assume that humans would produce canaline by dietary consumption of canavanine. Canaline is a natural product of C. ensiformis (9), and the unripened seed of Astragalus sinicus (10).
BIOLOGICAL EFFECTS
Incorporation of canaline into the artificial diet of the tobacco hornworm, Manduca sexta [Sphingidae] causes depauperate larvae, reduces successful larval-pupal ecdysis, increases the severity of pupal and adult malformation, decreases survival of all developmental stages, and significantly attenuates ovarial mass production (11). Injected into the adult moth, canaline elicits almost continuous motor activity. Initially, the insect flys correctly, but it quickly becomes disorientated; muscle activity is less patterned, even though wing movement continues. Neither axonal conduction of action potentials nor the activity of mechanoreceptors is affected, but the postsyntaptic potential of flight muscle fibers is prolonged; after 20 to 40 min the electrical activity of muscle fibers is normal. This neuropharmacological agent clearly affects central nervous system function, but its exact mode of action remains to be resolved (12).
ANTIMETABOLIC PROPERTIES
Typically, canaline is not an effective competitive inhibitor of ornithine (13); for example, canaline does not significantly inhibit ornithine carbamoyltransferase (EC 2.1.3.3) (14), but this enzyme can synthesize O-ureidohomoserime, the ureidooxy-containing counterpart of citrulline, from canaline (15). Canaline does not adversely affect ornithine metabolism in human liver (16), but it does disrupt vaccinia virus replication in HeLa cells. Attempts at reversing this adverse effect with ornithine were unsuccessful (17).
On the other hand, canaline's limited potential to antagonize ornithine metabolism is revealed by the finding that hog kidney transamidinase mediates transfer of an amidine moiety from donors such as arginine, canavanine, and guanidinoacetate to ornithine and canaline (18). Both recipient amino acids were metabolically active (19). This transamidination reaction made possible the first synthesis of L-[guanidinooxy-14C]canavanine by enzyme-directed transfer of carbon from L-[guanidino-14C]arginine to canaline (20).

Metabolism and chemical reactions involving L-canaline a. hydrolytic cleavage of L-canavanine employing arginase (EC 3.5.3.1) to generate L-canaline and urea (not shown), b. oxime formation with glyoxylic acid, c. deamination, decarboxylation and ring closure to produce 3-isoxazolidone, d. L-amino acid oxidase (EC 1.4.3.2)-mediated oxidation, e. NADPH-dependent reductive cleavage of L-canaline to yield L-homoserine and ammonia (not shown), f. biosynthetic reactions supporting sulfur-bearing amino acid formation as established by the plant research of J. Giovanelli and his associates, g. Iysine formation via 2,3-dihydropicolinic acid or meso-2,6-diaminopimelic acid.
Except for canaline catabolism to homoserine by Canavalia ensiformis, ornithine is an active substrate in all biochemical reactions in which canaline functions. This exception results from the prodigious formation of canaline by C. ensiformis as part of its mobilization of the nitrogen atoms of the guanidinooxy group of canavanine via arginase-directed urea formation [Scheme 1]. (21,22). Jack bean is a rich source of urease; this enzyme accelerates the production of ammoniacal nitrogen from canavanine-derived urea. Canaline is the toxic by-product of this reaction. Investigation of canaline detoxification in this legume led to the discovery of canaline reductase (EC 1.6.6.-) which catalyzes a NADPH-dependent reduction of L-canaline to L-homoserine and ammonia (23). This reductive deamination not only detoxifies canaline, but also contributes to its low natural abundance while providing a means of assimilating the third and final carbon atom of the guanidinooxy group of canavanine. In addition, homoserine formation conserves carbon skeleton by fostering the biosynthesis of essential sulfur-bearing amino acids (24,25).
OXIME FORMATION
Canaline is a powerful inhibitor of pyridoxal phosphate-dependent enzymes. Rahiala et al. (13) were the first to recognize a direct reaction between canaline and the vitamin B6 moiety of pyridoxal- 5'-phosphate-containing enzymes by postulating: "...that canaline probably inhibits pyridoxal phosphate-containing enzyme by its nonenzymic, irreversible and stoichiometric binding with pyridoxal phosphate." The suggestion of such a complex had its foundation in the experimental efforts of DaVanzo et al. (26) who noted that aminooxyacetic acid, structurally similar to canaline, formed an oxime with pyridoxal phosphate. Cooper (27) reported that canaline reacts rapidly with glyoxylate and less effectively with other 2-oxo acids to form stable oximes. He also observed that canaline was a substrate for L-amino acid oxidase to yield a cyclic, internal oxime, isoxazoline-3-carboxylate [Scheme 1]. Canaline-2-oxo acid oximes also reacted with this enzyme to produce isoxazoline-3-carboxylate, a 2-oxo acid, and ammonia (27). Isoxazoline-3-carboxylate occurs in the Jack bean, Canavalia ensiformis where it may form from canaline (28). Much of the canaline synthesized from canavanine stored in the seed of C. ensiformis is detoxified by oxime formation with glyoxylate (29).
The ability of canaline to inactivate an enzyme by forming a stable, covalently-linked oxime was demonstrated directly with L-[U-14C]canaline which reacted with ornithine aminotransferase (EC 2.6.1.13) of larval Manduca sexta to yield an enzyme-bound, radiolabeled canaline-pyridoxal phosphate oxime (30). The radiolabeled oxime has been isolated from canaline-treated enzyme. Dialysis of canaline-inactivated ornithine aminotransferase against free pyridoxal phosphate gradually reactivated the enzyme as the oxime was replaced by pyridoxal phosphate (30). McCormick and Snell (31), who found that the pyridoxal- canaline oxime was a strong inhibitor of pyridoxal phosphokinase, reported that the oxime was bound to the enzyme 100 to 1,000 times more strongly than pyridoxal. In addition, canaline's ability to form oximes means that canaline can scavenge such essential 2-oxo-containing metabolites as pyruvate, oxaloacetate, and 2-oxoglutarate to deplete tricarboxylic acid reserves and carbon skeleton required for amino acid synthesis.
CANALINE-MEDIATED ENZYME INHIBITION
A family of 10 structurally related analogs of L-canaline have been synthesized and evaluated for their inhibitory effect on pyridoxal phosphate-dependent alanine aminotransferase (EC 2.6.1.2) obtained from porcine heart (32). These canaline congeners were selected to examine the effects of changing the stereochemistry about the chiral center, varying the aliphatic chain length, removing the a-amino group and modifying the aminooxy moiety. L-Canaline was the most potent inhibitor of the tested compounds: after a 5 min exposure, 10-7 M canaline elicited a 55% reduction in aminotransferase activity (IC50 = 2.5 x 10-7 M). By comparison, the methyl and ethyl esters of canaline reduced the enzyme activity by only 8 and 6%, respectively. The L-enantiomer forms of the tested compounds were more inhibitory than their corresponding D-stereoisomer; however, the D-enantiomers, particularly D-canaline (IC50 = 1.8 x 10-6 M), were active. Shortening or lengthening the aliphatic chain, relative to canaline, significantly curtailed canaline-mediated loss in aminotransferase activity; in contrast, the absence of an alpha-amino group had little affect on this loss. N-methylation of the terminal aminooxy moiety abrogated virtually all inhibition of aminotransferase activity. Thus, the presence of an intact carboxyl group at the chiral center, maintenance of the aliphatic chain length, and particularly an aminooxy moiety are of paramount importance in the expression of canaline-mediated inhibition of glutamic-pyruvate aminotransferase (32).
L-Canaline is a strong inhibitor of sheep liver hydroxymethyltransferase (EC 2.1.2.1). Incubation of this enzyme with 100 micromolar or 200 micromolar L-canaline for 5 min at 37°C caused a 45 or 75% loss, respectively in enzymic activity, Interestingly, with this protein, a 5 min pretreatment with 100 micromolar pyridoxal phosphate completely protected the enzyme. Hydroxylamine and aminooxyacetic acid (IC50 = 2-3 micromolar) were more potent inhibitors of this catalyst than L-canaline (IC50 = ca. 100 micromolar) (33). This differential toxicity was thought to be due to steric hindrance at the active site due to canaline's larger size. It would be valuable, therefore, to examine the potency of L-2-amino-3-(aminooxy)propionate, the lower homolog of canaline, against this mammalian enzyme. This compound can be prepared by simple ring opening of L-cycloserine (34).
Bolkenius et al. (35) conducted a comparative investigation of rat liver ornithine aminotransferase inactivation by L-canaline and 5-fluoromethylornithine [2,5-diamino-6-fluoro-hexanoic acid]. L-Canaline irreversibly inhibited this enzyme, and it reacted faster with this protein than 5-fluoromethylornithine. The enzyme half life at infinite inhibitor concentration was 2.5 min for D,Lcanaline and 4 min with the halogenated compound. These workers made the important observation that pyridoxamine-containing enzyme, formed in the course of ornithine deamination, did not react with canaline. Regeneration of pyridoxal 5'-phosphate at the active site of the protein by treatment with 2-oxoglutarate, restored canaline-mediated inactivation of the enzyme (35).
L-Canaline decreased the aspartic acid content of tissues of the medulla oblongata of male Wistar rats, but it did not affect the evoked release of this nonprotein amino acid into these tissues. These workers postulated that canaline-mediated inhibition of ornithine aminotransferase decreased the glutamic acid pool which in turn suppressed aspartic acid biosynthesis (36).
In a similar vein, intraseptal injection of 100 log of L-canaline into male Sprague-Dawley rats caused a 90% decrease in the ornithine aminotransferase activity of the septum tissues evaluated from animals killed 1 h later (37). Under these experimental conditions, brain glutamic acid levels diminished by 27%. Examination of the time course for canaline-mediated inhibition (100 microg dose) revealed that this adverse effect occurred within 5 min. It is evident from these studies, that L-canaline is a particularly potent inhibitor of ornithine aminotransferase both in vitro and in vivo. One must assume that steric parameters foster interaction between the pyridoxal phosphate moiety of the active site of this protein and canaline.
Mokrasch's (38) examination of L-lysine transport in astrocytes and astrocytoma cells, obtained from neonatal rat brains, involved the first use of L-canaline as a Iysine antimetabolite. He demonstrated that L-canaline inhibited L-lysine flux competitively (Ki = 4.6 + 1.5 mM). Canaline, however, was far less potent than S-aminoethylcysteine (Ki = 0.67 + 0.09 mM). The higher homolog of L-canaline, L-2-amino-5-(aminooxy)pentanoic acid, may represent a highly active Iysine antagonist. This compound can be synthesized from the higher homolog of L-homoserine via the 5-methyl ester of L-glutamic acid (34).
Canaline's effect on another pyridoxal phosphate-containing enzyme, namely ornithine decarboxylase (EC. 4.1.1.17) has also been examined (Figure 1).
Inhibition of L-ornithine decarboxylase activity. The enzyme activity of the control samples was 8,345 pmoles mg protein -1 hr-1. . The various compounds, at the indicated concentration were incubated with the enzyme for I h prior to enzyme assay which was predicated upon capture of 14CO2 from 14C-labeled L-ornithine. L-Canaline ( double hatch marks) , D-canaline (hatch marks parallel), L-2- amino-5 - [N-methylaminooxy]pentanoic acid (open bar), L-2-amino-3-(aminooxy)propionic acid (hatch marks upper right), and L-2-arnino-5-(aminooxy)pentanoic acid (hatch marks upper left).
These unpublished data revealed a pronounced canaline-dependent inhibition of this enzyme. That the D-enantiomer was even more active than its naturally occurring stereoisomer was an unexpected and intriguing finding. In contrast, D-canaline was significantly less effective than L- canaline in inhibiting rat liver aminotransferase activity (35). Addition of a methyl group to the aminooxy moiety to generate L-2-amino-5-[N-methy-L-aminooxy]pentanoic acid, or reduction of the aliphatic chain by one methylene, as in L-2-amino-3-(aminooxy)propionic acid, caused a loss of nearly all of the inhibitory activity against ornithine decarboxylase when tested at a 5 micromolar concentration. (Figure 1). The higher homolog of L-canaline, L-2-amino-5-(aminooxy)pentanoic acid, retained about one-half of the ability of the canaline in diminishing decarboxylation of L-ornithine (Worthen et al., unpub. obser.). At increasingly greater levels of these test compounds, their deleterious effect was enhanced.
Since canaline forms from canavanine, and the former might contribute to canavanine's anticancer potency, the ability of a number of canaline congeners to curtail the growth of MIAPaCa-2, a human pancreatic carcinoma line, in culture was evaluated (39, 40). This cell line was selected because of our on-going studies of the antineoplastic activity of L-canavanine against human pancreatic carcinoma that employ MIAPaCa-2 cells. Addition of 2.5 mM L-canaline to the cell culture medium resulted in more than a 90% reduction in cell viability (Na Phuket et al., unpub. obser.). Interestingly, D-canaline is surprisingly active; under comparable conditions it caused more than an 80% reduction in cell viability (IC50 for D-canaline = 0.8 mM). When the concentration of D-canaline reaches 5.0 mM, it is as potent as the naturally occurring enantiomer (IC50 = 0.6 mM).
Various esters of canaline such as the methyl, ethyl, propyl, isopropyl, butyl, and octyl derivative exhibited marked growth-inhibiting activity against MIAPaCa-2 cells. The IC50 value for these alkyl esters was: 0.8, 1.5, 1.0, 0.8, 0.8, and 0.1 mM, respectively. Chain shortening and lengthening, relative to canaline, did not attenuate the loss in cell viability. In fact, the only structural modification that reduced the efficacy of canaline was methylation of the aminooxy moiety (Na Phuket et al., unpub. obser.). Studies of canaline's antineoplastic activity in an animal model have not been conducted, but canaline and its congeners merit further scrutiny as potential chemotherapeutic agents.
L-Canaline emerges as a highly toxic natural product that potentially could represent a highly effective, protective allelochemical against insects, predators, and pests; yet, its natural abundance in canavanine-containing legumes appears to be low. Detailed examination of the L-canaline content of the root, leaf, stem and cotyledons of C. ensiformis conducted in the author's laboratory revealed only trace levels of this toxic nonprotein amino acid (29). Canaline's ability to react facilely with the carbonyl group of an aldehyde or certain organic acids to generate an oxime, and its potential ability to inactivate irreversibly B6-containing enzyme, including many aminotransferases and decarboxylases, created a highly deleterious natural product, but one whose antimetabolic action could not be readily circumvented by a canaline-storing plant. As a result, I believe, selection pressures over evolutionary time did not favor the accumulation of this potent toxicant in spite of its marked potential to serve as a protective allelochemical.
REFERENCES
1. M. KITAGAWA, and J. YAMADA. J. Biochem. (Tokyo) 16 339-349 (1932).
2. M. DAMODARAN, AND K.G.A. NARAYANAN, Biochem. J. 34 1449-1459 (1940). 3. J.F. THOMPSON, In: The Biochemistry of Plants. A Comprehensive Treatise, Vol. 5, B.J. Miflin ed, 375-402, Academic Press, New York (1980).
4. J. SAWER, AND L. KAPLAN, Amer. Antiquity 34 417-424 (1969).
5. D.E. KAY, Food Legumes, London, UK (1978).
6. E.A. BELL, Biochem. J. 75 618-620 (1960).
7. S. NATELSON, J. Agric. Food Chem. 33 414-419 (1985).
8. D.A. THOMAS, and G.A. ROSENTHAL, Toxicology & Appl. Pharm. 91 406-414. (1987).
9. J. MIERSCH, Naturwissenschaften 54 169-170 (1967).
10. H. INATOMI, F. INUGAI, and T. MURAKAMI, Chem. Pharm. Bull. 16 2521 (1973).
11. G.A. ROSENTHAL, and D.L. DAHLMAN, Comp. Biochem. Physiol. 52A 105-108 (1975).
12. A.E. KAMMER, D.L. DAHLMAN, and G.A. ROSENTHAL, J. Exptl. Biol. 75 123-132 (1978).
3. E.-L. RAHIALA, M.K. KEKOMAKI, J. JANNE, A. RAINA, and N.C.R. RAIHA, Biochim. biophys. Acta 227 337-343 (1971).
14. M. KEKOMAKI, E.-L. RAHIALA, and N.C.R. RAIHA, 47 33-38 (1969).
15. D. O'NEAL, Plant Physiol. 55 975-977 (1975).
16. S. NATELSON, A. KOLLER, H.-Y. YSEND, and R.F. DODS, Clin. Chem. 23 960-966 (1977).
17. J.D. WILLIAMSON, and L.C. ARCHARD, J. Gen. Virol. 30 81-89 (1976).
18. S. RATNER, and O. ROCHOVANSKY, Arch. Biochem. Biophys. 63 277-295 (1956). 1
9. S. RATNER, and O. ROCHOVANSKY, Arch. Biochem. Biophys. 63 296-315 (1956).
20. C.C. ALLENDE, and J.E. ALLENDE,1. Biol. Chem. 239 1102-1106 (1964).
21. G.A. ROSENTHAL, M. BERGE, A. OZINSKAS, and C. H. HUGHES, J. Food Agr. Chem. 36 1159-1163 (1988).
22. G.A. ROSENTHAL, and M. BERGE, J. Food Agr. Chem. 37 591-595 (1989).
23. G.A. ROSENTHAL, Proc. Natl. Acad. Sci. 89 1780-1784 (1992).
24. J. GIOVANELLI, and S.H. MUDD, Biochem. Biophys. Res. Comm. 31 275-280 (1961).
25. J. GIOVANNELLI, S. H. MUDD, and A.H. DATKO, In: The Biochemistry of Plants. A Comprehensive Treatise. Vol. 5, B.J. Miflin ea., 454-506, Academic Press, New York (1980).
26. J.P. DAVANZO, R.J. MATTHEWS, G.A. YOUNG, and F. WINGERSONA, Toxicol. Appl. Pharmacol. 6 396-401 (1964)
27. A.J.L. COOPER, Arch. Biochem. Biophys. 233 603-610 (1984).
28. M. SUGII, H. MIURA, and K. NAGATA, Phytochemistry 20 451-453 (1981).
29. G.A. ROSENTHAL, M. BERGE, and J. BLEILER, Biochem. Syst. Ecol. 17 203-206. (1989).
30. G.A. ROSENTHAL, and D.L. DAHLMAN, J. Biol. Chem. 265 868-873. (1990).
31. D.B. MCCORMICK, and E.E. SNELL, J. Biol. Chem. 236 2085- (1961). 32. D. WORTHEN, D.K. RATLIFF, G.A. ROSENTHAL, L. TRIFONOV, and P.A. CROOKS, Chem.Res. Toxicol. in press (1996).
33. N. BASKARAN, V. PRAKASH, H.S. SAVITHRI, A.N. RADHAKRISHNAN, and N. APPAJI RAO,Biochemistry 28 9613-9617 (1989).
34. G.A. ROSENTHAL, D.L. DAHLMAN, P.A. CROOKS, S.N.A. PHUKET, and L.S. TRIFONOV, J.Food Agr. Chem. 43 2728-2734 (1995).
35. F.N. BOLKENIUS, B. KNODGEN, and N. SEILER, Biochem. J. 268 409-414 (1990)
36. T. KUBO, M. KIHARA, and Y. MISU, Naunyn-Schmiedeberg's Arch. Pharmcol. 341 221-224 (1990).
37. J.T. WROBLEWSKI, W.D. BLAKER, and J.L. MEEK, Brain Research 329 161-168 (1985).
38. L.C. MOKRASCH, Membr. Biochem. 7 249-258 (1987).
39. D.S. SWAFFAR, C.Y. ANG, P.B. DESAI, and G.A ROSENTHAL, Cancer Research 54 6045-6048 (1994).
40. D.S. SWAFFAR, Y.A. CHOO, P.B. DESAI, G.A. ROSENTHAL, D.A. THOMAS, P.A. CROOKS, and W.J. JOHN, Anti-Cancer Drugs 6 586-593 (1995).